Volume 30, Number 10—October 2024
Dispatch
Clustering of Polymorphic Membrane Protein E Clade in Chlamydia trachomatis Lineages from Men Who Have Sex with Men
Abstract
Several Chlamydia trachomatis lineages identified through outer membrane protein A genotyping or multilocus sequence typing have been circulating worldwide among men who have sex with men. In a study in Tokyo, Japan, we demonstrate that such lineages commonly belong to a specific polymorphic membrane protein E clade across genotypes.
Chlamydia trachomatis infection is the most common sexually transmitted infection (STI) worldwide. Because most infections are asymptomatic, sexual transmission generally occurs without notification. This aspect of transmission creates a risk for persistent undiagnosed C. trachomatis infection, which can lead to ascending infection in the female genital tract and result in serious conditions, such as pelvic inflammatory disease, ectopic pregnancy, and infertility.
The standard epidemiologic marker used for C. trachomatis genotyping is ompA, which encodes the major outer membrane protein. C. trachomatis is classified into 18 genotypes on the basis of ompA diversity, and the genotypes are further categorized into 3 groups on the basis of their predominant anatomic sites: ocular (A–C), urogenital and anorectal (D–K), and lymphogranuloma venereum (L1–L3). The molecular epidemiology of C. trachomatis is characterized by the predominance of ompA genotypes D, G, and J among men who have sex with men (MSM) in many countries (1–8). Multilocus sequence typing (MLST) has revealed that MSM-specific sequence types (STs) are present in these genotypes (5–8) and that those STs are distributed globally, suggesting the presence of specific international transmission networks among MSM. However, how the specific STs were selectively disseminated among MSM across several ompA genotypes or whether they have any shared underlying characteristics are unclear. The purpose of this study was to characterize the molecular epidemiology of C. trachomatis among MSM in Tokyo, Japan.
We focused on C. trachomatis polymorphic membrane protein (Pmp) variation, which is considered to play a key role in the initial infection process (9,10). Among 9 Pmp groups (PmpA–PmpH) PmpE is the most diverse, and a specific clade has been identified in rectal samples from MSM (11), suggesting a potential target for molecular epidemiologic studies of C. trachomatis.
The clinical specimens were collected from MSM at an outpatient clinic at the National Center for Global Health and Medicine in Tokyo that specializes in providing care for MSM. We collected 7,200 pharyngeal and 1,904 urogenital specimens during October 2018–March 2021, and collected 703 rectal specimens during April 2019–March 2021. The men were participants in an HIV-negative cohort study on implementation of preexposure prophylaxis. The specimen collection methods have been described previously (12). In addition, 200 urogenital specimens and 42 cervical specimens were collected as non-MSM samples from outpatients attending general clinics (not specifically for MSM) with urinary or genital tract infections during the same period. The major departments of those clinics were obstetrics and gynecology (clinic A), gastroenterology (clinic B), and urology (clinic C). We selected patients who had clinically suspected Neisseria gonorrhoeae or C. trachomatis infection. This study was approved by the ethics committee of the Tokyo Metropolitan Institute of Public Health (approval no. 3KENKENKENDAI465GOU).
We sequenced the C. trachomatis–positive specimens, confirmed using an Aptima Combo 2 transcription-mediated amplification test (Hologic, https://www.hologic.com), to determine the ompA genotypes, as described previously (13). We performed MLST targeting 5 regions (hctB, CT058, CT144, CT172, and pbpB) using the Uppsala scheme as described in the PubMLST website (https://pubmlst.org/organisms/chlamydiales-spp), assigning new STs when they were discovered. On the basis of the determined STs, we constructed a minimum-spanning tree using the GrapeTree tree visualization program (14) with the MSTreeV2 algorithm. We amplified the near–full length of PmpE-encoding regions (2740 bp), which includes 5 variable regions (11), by nested PCR using primer sets. We used pmpE_1st_F (5′-GAAAAAAGCGTTTTTCTTTTTCCTTATCG-3′) and pmpE_1st_R (5′-TCCCCATTGAGATAATTACAGAAGGTTGA-3′) for the first PCR and used pmpE_2nd_F (5′-AACTCAGTTCCAGATCCTACGAAAGAGTC-3′) and pmpE_2nd_R (5′-ACTGGAAATGGAGAGTTAACCAACTCAAAG-3′) for the second PCR. We sequenced the PCR products through amplicon sequencing using MiSeq (Illumina, https://www.illumina.com). We constructed a nonrooted phylogenetic tree with the neighbor-joining method on the basis of the amino acid differences with MEGA7 software (https://www.megasoftware.net) using the amino acid sequences (907 aa) obtained by computational translation of DNA sequences corresponding to nucleotide numbers 1,025,723–1,028,443 of the C. trachomatis D/UW-3/CX genome (AE001273) (15).
Figure 1

Figure 1. Minimum spanning tree based on sequence types (STs) and ompA of 298 Chlamydia trachomatis samples in study of clustering of specific polymorphic membrane protein E clade in ...
We fully analyzed a total of 298 C. trachomatis–positive specimens (245 from MSM and 53 from non-MSM) with ompA genotyping, MLST, and PmpE sequencing (Appendix 1). Although specimens were repeatedly collected from several MSM participants on different dates, no duplicate data (identical ST detection from the same site on different collection dates) were collected. The predominant ompA genotypes in the MSM population were D, G, and J (Figure 1; Appendix 2 Table 1), as previously reported in several countries (5–8). The most frequently detected STs were ST108 and its single-locus variants (SLVs) (e.g., ST33, ST52) and ST109 and its SLV (e.g., ST194). The main ompA genotypes were G/J in the ST108 lineage and D in the ST109 lineage (Appendix 2 Table 2). ST108 and ST109 are quadruple-locus variants of each other.
Figure 2
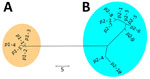
Figure 2. Nonrooted phylogenetic tree created on the basis of polymorphic membrane protein E of 298 Chlamydia trachomatis samples in study of clustering of specific polymorphic membrane protein E clade in ...
Figure 3

Figure 3. Minimum spanning tree based on sequence types (STs) and polymorphic membrane protein E (PmpE) of 298 Chlamydia trachomatis samples in study of clustering of specific PmpE clade in ...
We detected 15 PmpE sequences in the 298 samples (p1–1 to p1–5 and p2–1 to p2–10), and those were clearly separated into 2 clades (named as p1 and p2) reflecting the MSM and non-MSM populations (Figure 2; Appendix 2 Figure). A few MSM samples were classified as p2, whereas no non-MSM samples were classified as p1. In MSM, the prevalence of the p1 clade did not differ significantly in urogenital, pharyngeal, and rectal samples (p = 0.141 by Fisher exact test) (Table), suggesting that the difference in clade between MSM and non-MSM samples was not attributable to differences in the anatomic sample collection sites. To investigate the phylogenetic relationships between the PmpE clades, we created a minimum spanning tree from STs showing the relationship to p1 and p2 (Figure 3). We further divided both lineages into likely sublineages corresponding to the difference in the PmpE clades (Figures 1, 3).
Tokyo is a capital city with a population of >10 million and is connected to the 2 largest international airports in Japan; therefore, the similarity of the genotype distribution observed in this study to that observed in other countries is not surprising. The predominant C. trachomatis lineages among MSM in this study were centered around ST108 and ST109. Of the samples from MSM in this study, 89.4% (219/245) were major STs or their SLVs had been previously reported among MSM in other countries (5–8), demonstrating that the circulating lineages among MSM in Tokyo were typical of STs circulating internationally in MSM populations. In contrast, none of the STs of non-MSM samples were classified as major MSM STs, suggesting that the samples from non-MSM patients in this study were not linked to STs circulating in the global MSM population.
This study revealed that most MSM-associated C. trachomatis STs belonged to the specific PmpE clade p1. This finding indicates that nonsimplex C. trachomatis lineages with shared microbiological characteristics involved in the infection process (9,10) likely disseminated in parallel through international MSM networks and that those shared characteristics might be involved in the infection process and transmission. Taken together, this study demonstrates the importance of PmpE as a target for molecular epidemiologic investigation to clarify the dynamics of C. trachomatis transmission.
Ms. Mitobe is a pharmacist who works as a senior scientist at the Tokyo Metropolitan Institute of Public Health, Tokyo, Japan. Her primary research interest is sexually transmitted diseases.
Acknowledgments
We thank Shigenobu Nishijima, Shinsuke Watanabe, and Toshihisa Iwabuchi for collecting the clinical specimens.
This work was supported by the National Center for Global Health and Medicine (grant nos. 19A1002, 20A1020), JSPS KAKENHI (grant no. 24K10230), and the Takeda Science Foundation.
References
- Klint M, Löfdahl M, Ek C, Airell A, Berglund T, Herrmann B. Lymphogranuloma venereum prevalence in Sweden among men who have sex with men and characterization of Chlamydia trachomatis ompA genotypes. J Clin Microbiol. 2006;44:4066–71. DOIPubMedGoogle Scholar
- Twin J, Moore EE, Garland SM, Stevens MP, Fairley CK, Donovan B, et al. Chlamydia trachomatis genotypes among men who have sex with men in Australia. Sex Transm Dis. 2011;38:279–85. DOIPubMedGoogle Scholar
- Li JH, Cai YM, Yin YP, Hong FC, Shi MQ, Feng TJ, et al. Prevalence of anorectal Chlamydia trachomatis infection and its genotype distribution among men who have sex with men in Shenzhen, China. Jpn J Infect Dis. 2011;64:143–6. DOIPubMedGoogle Scholar
- Qin X, Zheng H, Xue Y, Ren X, Yang B, Huang J, et al. Prevalence of Chlamydia trachomatis genotypes in men who have sex with men and men who have sex with women using multilocus VNTR analysis—ompA typing in Guangzhou, China. PLoS One. 2016;11:
e0159658 . DOIPubMedGoogle Scholar - Piñeiro L, Villa L, Salmerón P, Maciá MD, Otero L, Vall-Mayans M, et al. Genetic characterization of non-lymphogranuloma venereum Chlamydia trachomatis indicates distinct infection transmission networks in Spain. Int J Mol Sci. 2023;24:6941. DOIPubMedGoogle Scholar
- Bom RJ, van der Helm JJ, Schim van der Loeff MF, van Rooijen MS, Heijman T, Matser A, et al. Distinct transmission networks of Chlamydia trachomatis in men who have sex with men and heterosexual adults in Amsterdam, The Netherlands. PLoS One. 2013;8:
e53869 . DOIPubMedGoogle Scholar - Herrmann B, Isaksson J, Ryberg M, Tångrot J, Saleh I, Versteeg B, et al. Global multilocus sequence type analysis of Chlamydia trachomatis strains from 16 countries. J Clin Microbiol. 2015;53:2172–9. DOIPubMedGoogle Scholar
- Versteeg B, Bruisten SM, van der Ende A, Pannekoek Y. Does typing of Chlamydia trachomatis using housekeeping multilocus sequence typing reveal different sexual networks among heterosexuals and men who have sex with men? BMC Infect Dis. 2016;16:162. DOIPubMedGoogle Scholar
- Debrine AM, Karplus PA, Rockey DD. A structural foundation for studying chlamydial polymorphic membrane proteins. Microbiol Spectr. 2023;11:
e0324223 . DOIPubMedGoogle Scholar - Favaroni A, Hegemann JH. Chlamydia trachomatis polymorphic membrane proteins (Pmps) form functional homomeric and heteromeric oligomers. Front Microbiol. 2021;12:
709724 . DOIPubMedGoogle Scholar - Suchland RJ, Carrell SJ, Ramsey SA, Hybiske K, Debrine AM, Sanchez J, et al. Genomic analysis of MSM rectal Chlamydia trachomatis isolates identifies predicted tissue-tropic lineages generated by intraspecies lateral gene transfer-mediated evolution. Infect Immun. 2022;90:
e0026522 . DOIPubMedGoogle Scholar - Mizushima D, Takano M, Uemura H, Yanagawa Y, Aoki T, Watanabe K, et al. High prevalence and incidence of rectal Chlamydia infection among men who have sex with men in Japan. PLoS One. 2019;14:
e0220072 . DOIPubMedGoogle Scholar - Yoshida H, Kishi Y, Shiga S, Inoue S, Hagiwara T. Serotyping of Chlamydia trachomatis by polymerase chain reaction [in Japanese]. Sex Transm Infect. 1995;6:40–5.
- Zhou Z, Alikhan NF, Sergeant MJ, Luhmann N, Vaz C, Francisco AP, et al. GrapeTree: visualization of core genomic relationships among 100,000 bacterial pathogens. Genome Res. 2018;28:1395–404. DOIPubMedGoogle Scholar
- Kumar S, Stecher G, Tamura K. MEGA7: Molecular Evolutionary Genetics Analysis version 7.0 for bigger datasets. Mol Biol Evol. 2016;33:1870–4. DOIPubMedGoogle Scholar
Figures
Table
Cite This ArticleOriginal Publication Date: September 18, 2024
Table of Contents – Volume 30, Number 10—October 2024
| EID Search Options |
|---|
|
|
|
|
|
|
Please use the form below to submit correspondence to the authors or contact them at the following address:
Hiroaki Kubota, 3-24-1 Hyakunincho, Shinjuku-ku, Tokyo, 169-0073 Japan
Top