Volume 19, Number 12—December 2013
Letter
Porcine Hokovirus in Domestic Pigs, Cameroon
Cornelia Adlhoch1 , Marco Kaiser, Manchang T. Kingsley, Norbert Georg Schwarz, Markus Ulrich, Vanessa S. de Paula, Julian Ehlers, Anna Löwa, Achukwi M. Daniel, Sven Poppert, Jonas Schmidt-Chanasit, and Heinz Ellerbrok
, Marco Kaiser, Manchang T. Kingsley, Norbert Georg Schwarz, Markus Ulrich, Vanessa S. de Paula, Julian Ehlers, Anna Löwa, Achukwi M. Daniel, Sven Poppert, Jonas Schmidt-Chanasit, and Heinz Ellerbrok
Figure
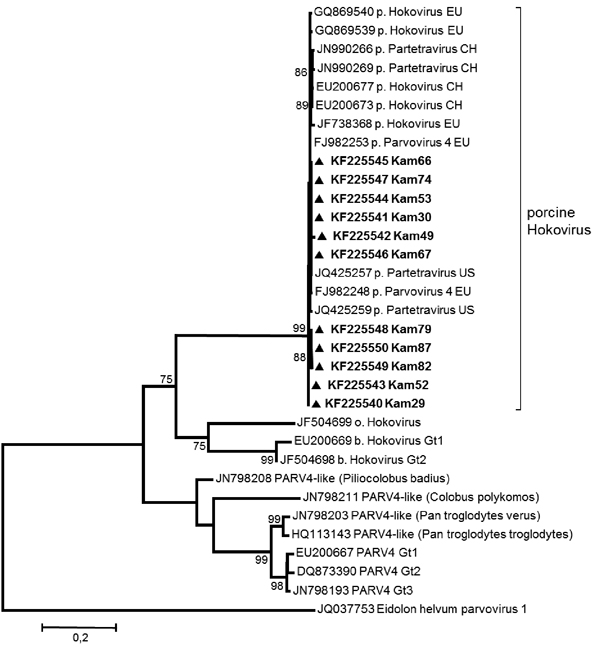
Figure. . Phylogenetic tree of near full-length (4.7 kbp) and partial sequences (open reading frame 2, 0.4–1.9 kbp) of porcine hokovirus (HoV)/partetraviruses (PtV) created by using MEGA5.1 (www.megasoftware.net) with the maximum-likelihood method (GTR+G+I) and bootstrap analysis of 500 resamplings. New sequences from Cameroon are shown in boldface. EU, Europe; CH, China; US, United States; Gt, genotype; PARV4, parvovirus 4. Scale bar indicates nucleotide substitutions per site.
1Current affiliation: European Centre for Disease Prevention and Control, Stockholm, Sweden.
Page created: November 20, 2013
Page updated: November 20, 2013
Page reviewed: November 20, 2013
The conclusions, findings, and opinions expressed by authors contributing to this journal do not necessarily reflect the official position of the U.S. Department of Health and Human Services, the Public Health Service, the Centers for Disease Control and Prevention, or the authors' affiliated institutions. Use of trade names is for identification only and does not imply endorsement by any of the groups named above.