Volume 30, Number 9—September 2024
Research
Autochthonous Leishmaniasis Caused by Leishmania tropica, Identified by Using Whole-Genome Sequencing, Sri Lanka
Abstract
Cutaneous leishmaniasis is atypical in Sri Lanka because Leishmania donovani, which typically causes visceral disease, is the causative agent. The origins of recently described hybrids between L. donovani and other Leishmania spp. usually responsible for cutaneous leishmaniasis remain unknown. Other endemic dermotropic Leishmania spp. have not been reported in Sri Lanka. Genome analysis of 27 clinical isolates from Sri Lanka and 32 Old World Leishmania spp. strains found 8 patient isolates clustered with L. tropica and 19 with L. donovani. The L. tropica isolates from Sri Lanka shared markers with strain LtK26 reported decades ago in India, indicating they were not products of recent interspecies hybridization. Because L. tropica was isolated from patients with leishmaniasis in Sri Lanka, our findings indicate L. donovani is not the only cause of cutaneous leishmaniasis in Sri Lanka and potentially explains a haplotype that led to interspecies dermotropic L. donovani hybrids.
Leishmaniasis is a heterogeneous group of diseases caused by protozoan parasites of the genus Leishmania, transmitted by the bite of phlebotomine sand flies (1). The 3 typical clinical presentations of leishmaniasis affecting humans are visceral leishmaniasis (VL), cutaneous leishmaniasis (CL), and mucocutaneous leishmaniasis (MCL) (1). Those who are most affected are persons in Asia, Africa, and Latin America who suffer from poverty (2).
Although Leishmania donovani causes VL in other countries in Asia and Africa, an atypical variant of the same species is almost exclusively associated with CL in Sri Lanka (3). Common symptoms of CL are papules, nodules, plaques, and ulcers. Leishmaniasis cases in Sri Lanka have increased over the past 2 decades, from 22 in 2001 to 3,389 in 2022 (https://www.epid.gov.lk). Over the past 2 decades, <10 cases of leishmaniasis have been VL in Sri Lanka; some cases have included severe chronic conditions preceding or occurring after the Leishmania infection (4). There are 2 primary hotspots in Sri Lanka, 1 in the southern province and 1 in the north-central province (5).
The genetic factors associated with disease phenotypes of leishmaniasis are not well understood. The A2 multigene family in Leishmania might be associated with either dermotropic or viscerotropic phenotypes (6). A2 gene variants encode stress-induced transmembrane proteins that are key for the parasite tropism to internal organs observed in visceralizing species (6). Abnormal changes in the number of chromosome copies that characterize extensive aneuploidy, usually harmful in most complex organisms, are frequent and highly tolerated in Leishmania parasites because they have a role in gene expression modulation (7,8).
The leishmaniasis disease profile worldwide is typically associated with the causative species of Leishmania (9). However, descriptions of emerging isolates and atypical phenotypes have made this association less clear (6,10). Recent genetic studies have shown that the Leishmania natural population structure is more complex than previously thought, partially because of the plasticity of the parasite genome and the occurrence of sexual reproduction (11). Isolates from the same clade might carry relevant genetic features accounting for shifts in clinical phenotypes. For example, L. donovani isotype MON-37 (from the Montpellier typing system) is the known causative agent of CL in Sri Lanka, and MON-2 is the known causative agent of VL in India (3,12).
A more recent analysis of L. donovani clinical isolates from Sri Lanka reported interspecies genomic hybrids between L. donovani and 2 common cutaneous species, L. major from Africa and L. tropica from the Middle East (13). The evidence of hybridization and introgression history in the Leishmania population in Sri Lanka suggests genetic exchange might have played a role in the insurgence of dermotropic L. donovani. However, many epidemiologic aspects of this model are unclear. Most CL causing L. donovani isolates described to date do not display clear evidence of hybridization with dermotropic species, and the parental strains of possible hybrid parasites in Sri Lanka remain unknown. Currently, <20 high quality next-generation sequencing (NGS) datasets of Leishmania spp. isolated from patients in Sri Lanka are publicly available (13). Therefore, it is crucial to perform whole-genome sequencing (WGS) analysis on a wider range of clinical isolates to better understand the atypical pathogenesis of leishmaniasis in Sri Lanka.
We studied WGS results of 27 Leishmania clinical isolates from Sri Lanka and made multiple genetic comparisons by using 32 different Old World Leishmania strains, including 5 interspecies L. donovani hybrids previously reported in Sri Lanka. Among the genomes analyzed, we describe autochthonous L. tropica isolates in patients from Sri Lanka who, apart from 1 exception, did not have travel history outside the country.
This study has been approved by the Ethics Review Committee, Faculty of Medicine, University of Colombo, Sri Lanka (approval no. EC-16-080). Written, informed consent was obtained from the participants.
Leishmania Culture
We cultured Leishmania promastigotes from 27 lesion aspirates (26 CL and 1 MCL) in M199 culture medium supplemented with 10% heat-inactivated fetal bovine serum (Thermo Fisher Scientific, https://www.thermofisher.com) and 100 IU/ml of penicillin and 100mg/ml of streptomycin (Thermo Fisher Scientific). We incubated the cultures at 25 ± 1°C until the logarithmic phase promastigote count reached 107/mL and then harvested them.
Genomic DNA Isolation and Sequencing
We extracted DNA from the cultured Leishmania promastigotes by using a QIAamp DNA Mini Kit (QIAGEN, https://www.qiagen.com) and prepared libraries by using the Nextera XT DNA library preparation kit (Illumina, https://www.illumina.com) and the Nextera DNA Flex library preparation kit (Illumina), according to the manufacturer’s instructions. Paired-end sequencing was conducted by Applied Biological Materials (British Columbia, Canada) by using the NextSeq (2×75bp) and HiSeq 4000 (2×150bp) platforms (Illumina).
WGS Data Analysis
We analyzed WGS data of 27 clinical isolates from this study and 32 Leishmania genomes of Old World species available from the National Center for Biotcechnology Information Sequence Read Archive (https://www.ncbi.nlm.nih.gov/sra) or the European Nucleotide Archive (https://ebi.ac.uk/ena). We detected genome-wide single-nucleotide polymorphisms (SNPs) in each sample after mapping high-quality reads to the reference strain, L. tropica L590 or L. donovani CL–Sri Lanka (genome available on TriTrypDB, https://tritrypdb.org). We extracted the consensus sequences of 7 different genetic markers to identify the clinical isolates at the species level. Phylogenetic analysis by NGS multilocus sequence typing (MLST) involved 59 genomes of nucleotide sequences with the indicated number of base pairs from the following genes: ribosomal RNA internal transcribed spacer (ITS; L. donovani CL–Sri Lanka CP029526:1015798–1016063), 265 bp; glucose-6-phosphate dehydrogenase (G6PD; LtrL590_34:26953–27953), 1,001 bp; glucose-6-phosphate isomerase (GPI; LtrL590_12:302579–303479), 901 bp; isocitrate dehydrogenase precursor (ICD; LTRL590_SCAF000112:124407–125407), 1,001 bp; phosphomannose isomerase (PMI; LtrL590_32:632360–633360), 1,001 bp; aspartate aminotransferase (AST; LtrL590_35:290874–291838), 965 bp; and inosine-guanine nucleoside hydrolase (IGNH; LtrL590_14:44174–44933), 760 bp (14). We extracted the ITS reference sequence from the L. donovani CL–Sri Lanka reference genome because the ITS sequence is not fully assembled in L. tropica L590. We conducted phylogenetic analysis with maximum-likelihood method and tamura-nei model by using NGS MLST sequences as input in MEGA X (15). We compiled the pileup of read alignments to each locus used for 3 representative L. tropica genomes (Appendix 1 Figure 1). We checked the aneuploidy profiles by inferring the chromosome copy values from whole-genome analysis normalized read depth after we mapped to the L. tropica L590 reference genome v.57, found in TriTrypDB, and assuming that all samples are diploid.
To confirm the absence of species admixture in the new L. tropica from Sri Lanka, we studied the genomewide zygosity profiles and the population genetic structure by tracking SNP frequencies in all 59 WGS samples. After mapping, we used aligned reads to identify SNPs in heterozygosity (allele frequency 0.15–0.85) or in homozygosity (allele frequency >0.85) by using the PAINT software suite (16). We investigated the gene sequences of the A2 virulence factor (L. donovani CL–Sri Lanka CP029521:270000–320000) to identify any association with the phenotype of CL. We generated pseudo-sequences from 1,368,585 high-quality whole-genome linked SNPs that had information in >50% of the samples to build phylogenetic network trees in SplitsTree v.6.0.2.3 (https://software-ab.cs.uni-tuebingen.de/download/splitstree6/welcome.html) (17). We calculated hamming distances and used the neighbor-net method to generate a splits network. A total of 1,000 bootstrap split replicates were used for internode confirmation of the phylogenetic tree of the L. tropica genomes, with a minimum 50% cutoff.
Data Availability
Data supporting the conclusions of this article are within the article and Appendix files, including Sequence Read Archive accession numbers for all the genomic data used in this work (Appendix 2 Tables 1–3). We deposited raw sequencing data in the National Center for Biotechnology Information BioProject archive (accession no. PRJNA904745).
Figure 1

Figure 1. Geographic locations of patients with cutaneous or mucocutaneous leishmaniasis in Sri Lanka. The mucocutaneous leishmaniasis patient was from the Puttalam district (A), Anamaduwa subdistrict. The cutaneous leishmaniasis patients were from...
We recovered Leishmania clinical isolates from 27 patients in 3 administrative districts in Sri Lanka: Hambantota (n = 25), Matara (n = 1), and Puttalam (n = 1) (Figure 1). The CL patients were from the southern region and the MCL patient was from the north-western region of the country. Clinical manifestations of the CL lesions consisted of 8 papules, 3 plaques, and 15 ulcers. Eight of the ulcers were ulcerated nodules (Appendix 2 Table 4).
Species Variability among Clinical Isolates
Figure 2

Figure 2. Phylogenetic analysis of Leishmania spp. clinical patient isolates from Sri Lanka and reference Leishmaniaspp. strains using sequences of the ribosomal RNA internal transcribed spacer (ITS). Sri...
NGS and MLST revealed that 19 genomes sequenced in this study belonged to the L. donovani complex and 8 to L. tropica. This finding was observed in every single locus investigated by MLST (Figure 2; Appendix 1 Figures 2, 3). Of note, the ICD gene sequence of the single MCL isolate, HS1, is more similar to L. tropica strains from the Middle East (including LtKubba from Syria, and LtLT1 and LtLT2 from Lebanon) than to isolates from Sri Lanka (Figure 2) (8,18).
Chromosome Copy Profiles
Figure 3

Figure 3. Highly conserved aneuploidy profiles within the cutaneous Leishmania tropica clade from Sri Lanka. A) Heat map visualization of Leishmaniaabnormal chromosome numbers in 27 patient isolates from...
Genome-wide read depth analysis revealed highly conserved chromosome copy profiles among the 7 Sri Lanka L. tropica CL isolates (H9, H15, H19, H21, H24, H27, and H34), and a divergent pattern in the single MCL isolate, HS1 (Figure 3, panels A, B). Polysomy of chromosome 31 (>3 copies) was seen in all samples. This polysomy was commonly observed in previous studies on Leishmania spp (19,20).
Trisomy (2.4–3.5 copies) of chromosomes 2, 21, and 31 and near-trisomy (2.2–2.4 copies) of chromosomes 5, 8, 10, and 13 were detected in L. tropica isolates from Sri Lanka except for HS1, which displayed trisomic chromosomes 6, 9, 11, 14, 19, 20, 30, 32, and 33 (Figure 3, panel B). In comparison, L. donovani clinical isolates from Sri Lanka displayed a more heterogenous distribution of chromosome copy values with varied karyotypes (Figure 3, panels A, B). Of the 3 previously reported L. donovani–L. tropica hybrids, SRR67 and SRR66 showed multiple near-trisomy matches with the L. tropica isolates from Sri Lanka on chromosomes 5, 8, 10, and 13. Of note, near-monosomy of chromosome 2 (1.2–1.4 copies) was detected in SRR64, SRR65, and SRR69 (Figure 3, panel B).
Genome-Wide Zygosity Profiles and Phylogenetic Network Analysis
Figure 4

Figure 4. Frequency of genomewide heterozygous and homozygous SNPs in all genomes analyzed and presented as percent stacked bars after mapping sequencing reads from Leishmaniaspp. isolates from Sri Lanka. A)...
The total number of SNPs per sample ranged from 117,729 (in LtMA37 WGS) to 2,059,343 (in SRR65 WGS) within the sample cohort after mapping reads to the L. tropica L590 reference genome (Figure 4 panel A). The 3 previously reported L. donovani–L. tropica hybrids showed low homozygosity (<0.12; SRR66–69). In contrast, most of the SNPs in the L. donovani isolates from Sri Lanka (H105–165) and in the intraspecies L. donovani hybrid LdHPCL66, identified in the Himachal Pradesh province of India, were homozygous (>0.98) (Figure 4, panel A) (10). Highly skewed heterozygosity was detected in most of the L. tropica genomes because they are genetically more similar to the L. tropica L590 reference genome than to L. donovani. For comparison, we also mapped reads to the L. donovani CL–Sri Lanka reference genome (Figure 4, panel B). As we expected for L. donovani–L. tropica progeny, both the genome-wide SNP frequencies and absolute numbers in the previously reported hybrids SRR66–69 were largely unaffected by the reference genome used for mapping (Figure 4). All L. donovani laboratory strains showed high ratios of homozygous SNPs.
Figure 5
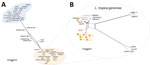
Figure 5. Phylogenetic network of all Leishmania isolates from Sri Lanka and Old World strains analyzed and visualized as a splits tree built using genomewide single-nucleotide polymorphisms in SplitsTree 6 (...
We generated a network splits tree by using whole-genome SNPs for all the 59 Leishmania genomes, highlighting the separation between the L. donovani and L.tropica isolates from Sri Lanka, with the hybrids found in the middle point of the tree (Figure 5, panel A). A more detailed analysis by using only the data from the L. tropica genomes revealed 2 different subclusters of L. tropica in Sri Lanka. Although HS1 co-clusters with strains found in Syria (LtKubba) (8) and Lebanon (LtLT1 and LtLT2) (18) (Appendix 1 Figure 4, panels A, B), the other L. tropica from Sri Lanka form a discrete subgroup more genetically similar to the LtK26 strain from India (Figure 5, panel B). This finding corroborated the phylogenetic profile observed by the MLST analysis by using the ICD gene sequence (Appendix 1 Figure 3, panel C). Identification of L. tropica phylogenetic groups confirms worldwide L. tropica populations described by a previous comparative genomics study (18). We further expand on those findings by suggesting an additional population, consisting of LtK26 strain from India (8) and the H9–34 L. tropica from Sri Lanka.
Genomewide Genetic Variations and Shared Ancestries
Figure 6
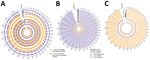
Figure 7

Figure 7. Representative nucleotide-level visualization of the inheritance of parental allelic markers on chromosomes 1 (upper panel) and 36 (lower panel) in L. donovani–L. tropica hybrid SRR66, ...
From the list of SNPs identified by using the PAINT software suite (16), we selected the 1,354,425 homozygous alleles that were different between L. tropica K26 and L. donovani SL2706 as representatives of the 2 clades found in Sri Lanka. We tracked their inheritance in hybrids SRR66, SRR67, and SRR69 and in the 27 Sri Lanka isolates. L. donovani–L. tropica markers were remarkably heterozygous across the whole genome of the SRR66–69 hybrids, suggesting they have undergone recent hybridization with biparental contribution from L. donovani and L. tropica in all 36 chromosomes (Figure 6, panel A). The number of SNPs shared with the L. tropica lineage from Sri Lanka was lower in the L. donovani–L. major hybrids (SRR64–65) (Appendix 1 Figure 5); only 31.5% of the heterozygous SNPs were explained by the tested L. donovani–L. tropica admixture compared with 70% in the L. donovani–L. tropica hybrids (SRR66–69). Visual inspection at the single-nucleotide level confirmed the extensive heterozygosity of L. donovani–L. tropica markers in the SRR66–69 hybrids for most of the sites analyzed, which was not as evident in the SRR64–65 hybrids (Figure 7; Appendix 1 Figure 6). The L. donovani (H105-H165) isolates from Sri Lanka showed low heterozygosity of selected parental markers and high similarity, and the L. tropica (HS1, H9-H34) isolates from Sri Lanka showed low heterozygosity of selected parental markers and high similarity to L. tropica from India lineages, evidence that these isolates have not experienced recent admixture of species (Figure 6, panels B, C).
A2 Gene Variations
Figure 8

Figure 8. Coverage plots of whole-genome sequencing reads mapped to the reference strain L. donovanicutaneous leishmaniasis from Sri Lanka. A) The absence of reads on the A2 gene locus in...
L. donovani and the hybrids from Sri Lanka contain the full A2 gene sequences of all 4 annotated copies in the L. donovani CL–Sri Lanka reference genome on chromosome 22. Read depths in L. tropica isolates from Sri Lanka were virtually zero in those positions (Figure 8, panel A), except for HS1, which contained very low partial coverage of the A2 gene, indicating a truncated gene sequence. Of note, isolates H106, H109, and H119 causing plaque lesions in patients were the only isolates from Sri Lanka to carry A2 sequences that are highly similar to the L. donovani CL–Sri Lanka reference, which also induced a plaque lesion phenotype (Figure 8, panel B).
This study identified an endemic L. tropica causing leishmaniasis in Sri Lanka. The 7 patients with CL caused by L. tropica had never traveled overseas, and 1 had not traveled outside their district of residence for 5 years. The CL patients had been living in their area of residence either since birth or for 20–60 years. Thus, it is likely that these are autochthonous cases of CL caused by L. tropica.
This study identified MCL caused by the L. tropica isolate HS1 in Sri Lanka. The patient’s upper lip, nose, and the perinasal regions were affected similar to previously reported cases of MCL caused by L. tropica (25,26). This MCL patient had traveled to Dubai and Iraq 8 years before the diagnosis and had no history of cutaneous leishmaniasis. The time elapsed since the first appearance of symptoms to diagnosis was ≈8 months (Appendix 2 Table 4). Those facts suggest that either the patient began experiencing symptoms 7 years after a possible exposure during overseas travel or they acquired the infection in Sri Lanka.
The 7 cutaneous L. tropica isolates from Sri Lanka (H9–H34) were collected in southern Sri Lanka and belong to a genetically conserved subgroup that only co-clustered with L. tropica LtK26 from India. In contrast, the MCL-associated HS1 strain was isolated from a different geographic area and displayed a divergent chromosome copy profile more similar to L. tropica strains from Syria (LtKubba) and Lebanon (LtLT1 and LtLT2) than to the other L. tropica isolates from Sri Lanka (Figure 3; Figure 5, panel B), and a divergent genomewide SNP profile. Thus, it is possible that the infection was acquired in the Gulf region, and the mucosal manifestations might have become apparent years after the primary exposure.
Chromosome aneuploidy was evident among the study samples and the pattern differed between the L. tropica and L. donovani isolates. Noninteger chromosome copy values observed in this study are commonly found in Leishmania because of mosaic aneuploidy occurring in cultured parasite populations (19). Recent studies have shown that clonal growth in both sand flies and in vitro culture can by itself lead to major changes in chromosome copy values and karyotype diversity even after just 1 passage through the insect vector (20,27). In fact, concerning chromosome copy values, the L. donovani–L. tropica hybrids were among the most homogeneous genomes in our analysis.
From the data on patients’ residence and travel within Sri Lanka (Appendix 2 Table 4), it is evident that both L. tropica and L. donovani may not be restricted to a single administrative area at the micro level. Considering that L. tropica is predominantly causing anthroponotic leishmaniasis and that the patients with CL have traveled to other districts, further studies in additional locations and with other potential vectors would benefit the current understanding of leishmaniasis spread and genetic variability in Sri Lanka. In addition, zoonotic transmission of L. tropica through reservoirs such as rock hyraxes has been reported in other countries, and studies are needed to assess potential animal reservoirs of these parasites in Sri Lanka (28). Currently, there are no data available to dismiss either scenario, and both may represent valid and relevant routes of infection in the country. In addition to L. tropica causing cutaneous disease, its potential to visceralize in humans is known (29–31). Even though CL is the current predominant phenotype of leishmaniasis in Sri Lanka, the presence of L. tropica and L. donovani, both with potential visceralizing properties, raises concerns.
Studies on A2 gene expression in old world Leishmania spp. suggest its useful in determining species-specific organ tropism (32). Even though the pathogenicity of leishmaniasis is not fully understood, the understanding of CL lesions is they progress from an initial papule to a nodule or a plaque which can ultimately ulcerate. Several virulence factors were studied in the sequenced samples to understand the variability in Leishmania pathogenesis leading to different phenotypes. Because primarily cutaneous parasite species have a truncated A2 pseudogene, A2 is a gene family that occurs in L. donovani but not in L. tropica (6), a result seen in our study samples as well. A2 is necessary in the pathology of leishmaniasis and is known to aid in visceralization of the infection and survival within the host (33–35). Our analysis of the A2 gene in different phenotypes of L. donovani revealed marked differences in the plaque phenotype when compared with papules, nodules, and ulcers. A study conducted in Pakistan found that the clinical appearance of CL is not solely dependent on L. tropica genetic variations (36). This possibility could not be investigated in our study because most of the L. tropica–induced CL cases were ulcerated.
We have found there are >5 different populations of Leishmania in Sri Lanka: SL1, SL2A, SL2B, SL3 (13), and L. tropica from Sri Lanka (SL4). Our study has shown L. tropica has caused both CL and MCL in Sri Lanka. Those findings suggest further studies on differences in clinical phenotypes, potential vector species, and reservoirs of Leishmania species in Sri Lanka would be beneficial. Revisiting diagnostic and research approaches might be necessary in consideration of this species variability. Analysis by PCR and restriction fragment length polymorphism represent the inexpensive and easy laboratory methods available for distinguishing between the 2 species (37).
In conclusion, although this large Leishmania WGS dataset provides valuable insights, further whole-genome studies might lead to better understanding of the Leishmania species infecting people in Sri Lanka and might shed light on the extent of genetic heterogeneity of infective clades circulating in the country. This discovery is a turning point in understanding the pathology of leishmaniasis in Sri Lanka and may contribute to the development of more efficient diagnostics and treatments for CL and MCL caused by L. tropica.
Dr. Silva is a senior lecturer in parasitology at the University of Colombo, Sri Lanka. Her research interests include epidemiology, pathogenesis, and treatment and drug resistance of human parasites.
Acknowledgments
We thank the staff members of the study sites, the Department of Parasitology at the University of Colombo, and Hernan A. Lorenzi for support with programming code.
This study was financially supported by the University of Colombo (grant no. AP/3/2/2017/PG/31); the United States-Japan Cooperative Medical Sciences Program, National Institutes of Health, USA (award no. DAA3-19-65623); National Institute of Allergy and Infectious Diseases, National Institutes of Health (award no. U01AI136033); and in part by the Intramural Research Program, National Institute of Allergy and Infectious Diseases, National Institutes of Health.
Author contributions: study design and leadership, H.S., N.V.C., and N.D.K.; funding acquisition, H.S. and N.D.K.; study oversight and supervision, N.D.K., N.V.C., and D.L.S.; data and sample collection, H.S.; bioinformatics analysis, H.S., K.M., S.S., and T.R.F.; data processing and data visualization, T.R.F.; data interpretation and manuscript drafting, H.S. and T.R.F.; provision of bioinformatic scripts and pipelines, E.V.C.A.F. and M.E.G. All authors reviewed the manuscript.
References
- Desjeux P. Leishmaniasis: current situation and new perspectives. Comp Immunol Microbiol Infect Dis. 2004;27:305–18. DOIPubMedGoogle Scholar
- Alvar J, Yactayo S, Bern C. Leishmaniasis and poverty. Trends Parasitol. 2006;22:552–7. DOIPubMedGoogle Scholar
- Karunaweera ND, Pratlong F, Siriwardane HVYD, Ihalamulla RL, Dedet JP. Sri Lankan cutaneous leishmaniasis is caused by Leishmania donovani zymodeme MON-37. Trans R Soc Trop Med Hyg. 2003;97:380–1. DOIPubMedGoogle Scholar
- Siriwardana HVYD, Karunanayake P, Goonerathne L, Karunaweera ND. Emergence of visceral leishmaniasis in Sri Lanka: a newly established health threat. Pathog Glob Health. 2017;111:317–26. DOIPubMedGoogle Scholar
- Karunaweera ND, Senanayake S, Ginige S, Silva H, Manamperi N, Samaranayake N, et al. Spatiotemporal distribution of cutaneous leishmaniasis in Sri Lanka and future case burden estimates. PLoS Negl Trop Dis. 2021;15:
e0009346 . DOIPubMedGoogle Scholar - Zhang WW, Ramasamy G, McCall LI, Haydock A, Ranasinghe S, Abeygunasekara P, et al. Genetic analysis of Leishmania donovani tropism using a naturally attenuated cutaneous strain. PLoS Pathog. 2014;10:
e1004244 . DOIPubMedGoogle Scholar - Sterkers Y, Lachaud L, Crobu L, Bastien P, Pagès M. FISH analysis reveals aneuploidy and continual generation of chromosomal mosaicism in Leishmania major. Cell Microbiol. 2011;13:274–83. DOIPubMedGoogle Scholar
- Iantorno SA, Durrant C, Khan A, Sanders MJ, Beverley SM, Warren WC, et al. Gene expression in Leishmania is regulated predominantly by gene dosage. MBio. 2017;8:1–20. DOIPubMedGoogle Scholar
- Lypaczewski P, Thakur L, Jain A, Kumari S, Paulini K, Matlashewski G, et al. An intraspecies Leishmania donovani hybrid from the Indian subcontinent is associated with an atypical phenotype of cutaneous disease. iScience. 2022;25:
103802 . DOIPubMedGoogle Scholar - Inbar E, Shaik J, Iantorno SA, Romano A, Nzelu CO, Owens K, et al. Whole genome sequencing of experimental hybrids supports meiosis-like sexual recombination in Leishmania. PLoS Genet. 2019;15:
e1008042 . DOIPubMedGoogle Scholar - Karunaweera ND. Leishmania donovani causing cutaneous leishmaniasis in Sri Lanka: a wolf in sheep’s clothing? Trends Parasitol. 2009;25:458–63. DOIPubMedGoogle Scholar
- Lypaczewski P, Matlashewski G. Leishmania donovani hybridisation and introgression in nature: a comparative genomic investigation. Lancet Microbe. 2021;2:e250–8. DOIPubMedGoogle Scholar
- Zemanová E, Jirků M, Mauricio IL, Horák A, Miles MA, Lukeš J. The Leishmania donovani complex: genotypes of five metabolic enzymes (ICD, ME, MPI, G6PDH, and FH), new targets for multilocus sequence typing. Int J Parasitol. 2007;37:149–60. DOIPubMedGoogle Scholar
- Kumar S, Stecher G, Li M, Knyaz C, Tamura K. MEGA X: Molecular evolutionary genetics analysis across computing platforms. Mol Biol Evol. 2018;35:1547–9. DOIPubMedGoogle Scholar
- Shaik JS, Dobson DE, Sacks DL, Beverley SM. Leishmania sexual reproductive strategies as resolved through computational methods designed for aneuploid genomes. Genes (Basel). 2021;12:1–16. DOIPubMedGoogle Scholar
- Huson DH, Bryant D. Application of phylogenetic networks in evolutionary studies. Mol Biol Evol. 2006;23:254–67. DOIPubMedGoogle Scholar
- Salloum T, Moussa R, Rahy R, Al Deek J, Khalifeh I, El Hajj R, et al. Expanded genome-wide comparisons give novel insights into population structure and genetic heterogeneity of Leishmania tropica complex. PLoS Negl Trop Dis. 2020;14:
e0008684 . DOIPubMedGoogle Scholar - Dumetz F, Imamura H, Sanders M, Seblova V, Myskova J, Pescher P, et al. Modulation of aneuploidy in Leishmania donovani during adaptation to different in vitro and in vivo environments and its impact on gene expression. MBio. 2017;8:1–14. DOIPubMedGoogle Scholar
- Ferreira TR, Inbar E, Shaik J, Jeffrey BM, Ghosh K, Dobson DE, et al. Self-Hybridization in Leishmania major. MBio. 2022;13:
e0285822 . DOIPubMedGoogle Scholar - Bussotti G, Gouzelou E, Côrtes Boité M, Kherachi I, Harrat Z, Eddaikra N, et al. Leishmania genome dynamics during environmental adaptation reveal strain-specific differences in gene copy number variation, karyotype instability, and telomeric amplification. MBio. 2018;9:1–18. DOIPubMedGoogle Scholar
- Krzywinski M, Schein J, Birol I, Connors J, Gascoyne R, Horsman D, et al. Circos: an information aesthetic for comparative genomics. Genome Res. 2009;19:1639–45. DOIPubMedGoogle Scholar
- Samarasinghe SR, Samaranayake N, Kariyawasam UL, Siriwardana YD, Imamura H, Karunaweera ND. Genomic insights into virulence mechanisms of Leishmania donovani: evidence from an atypical strain. BMC Genomics. 2018;19:843. DOIPubMedGoogle Scholar
- Robinson JT, Thorvaldsdóttir H, Winckler W, Guttman M, Lander ES, Getz G, et al. Integrative genomics viewer. Nat Biotechnol. 2011;29:24–6. DOIPubMedGoogle Scholar
- Shirian S, Oryan A, Hatam GR, Daneshbod Y. Three Leishmania/L. species—L. infantum, L. major, L. tropica—as causative agents of mucosal leishmaniasis in Iran. Pathog Glob Health. 2013;107:267–72. DOIPubMedGoogle Scholar
- Shirian S, Oryan A, Hatam GR, Daneshbod K, Daneshbod Y. Molecular diagnosis and species identification of mucosal leishmaniasis in Iran and correlation with cytological findings. Acta Cytol. 2012;56:304–9. DOIPubMedGoogle Scholar
- Bussotti G, Li B, Pescher P, Vojtkova B, Louradour I, Pruzinova K, et al. Leishmania allelic selection during experimental sand fly infection correlates with mutational signatures of oxidative DNA damage. Proc Natl Acad Sci U S A. 2023;120:
e2220828120 . DOIPubMedGoogle Scholar - Waitz Y, Paz S, Meir D, Malkinson D. Effects of land use type, spatial patterns and host presence on Leishmania tropica vectors activity. Parasit Vectors. 2019;12:320. DOIPubMedGoogle Scholar
- Sohrabi Y, Havelková H, Kobets T, Šíma M, Volkova V, Grekov I, et al. Mapping the genes for susceptibility and response to Leishmania tropica in mouse. PLoS Negl Trop Dis. 2013;7:
e2282 . DOIPubMedGoogle Scholar - Magill AJ, Grögl M, Gasser RA Jr, Sun W, Oster CN. Visceral infection caused by Leishmania tropica in veterans of Operation Desert Storm. N Engl J Med. 1993;328:1383–7. DOIPubMedGoogle Scholar
- Sacks DL, Kenney RT, Kreutzer RD, Jaffe CL, Gupta AK, Sharma MC, et al. Indian kala-azar caused by Leishmania tropica. Lancet. 1995;345:959–61. DOIPubMedGoogle Scholar
- Ghedin E, Zhang WW, Charest H, Sundar S, Kenney RT, Matlashewski G. Antibody response against a Leishmania donovani amastigote-stage-specific protein in patients with visceral leishmaniasis. Clin Diagn Lab Immunol. 1997;4:530–5. DOIPubMedGoogle Scholar
- Zhang WW, Matlashewski G. Characterization of the A2-A2rel gene cluster in Leishmania donovani: involvement of A2 in visceralization during infection. Mol Microbiol. 2001;39:935–48. DOIPubMedGoogle Scholar
- McCall LI, Matlashewski G. Localization and induction of the A2 virulence factor in Leishmania: evidence that A2 is a stress response protein. Mol Microbiol. 2010;77:518–30. DOIPubMedGoogle Scholar
- Al-Khalaifah HS. Major molecular factors related to Leishmania pathogenicity. Front Immunol. 2022;13:
847797 . DOIPubMedGoogle Scholar - Khan NH, Llewellyn MS, Schönian G, Sutherland CJ. Variability of cutaneous leishmaniasis lesions is not associated with genetic diversity of Leishmania tropica in Khyber Pakhtunkhwa province of Pakistan. Am J Trop Med Hyg. 2017;97:1489–97. DOIPubMedGoogle Scholar
- De Silva NL, De Silva VNH, Deerasinghe ATH, Rathnapala UL, Itoh M, Takagi H, et al. Development of a highly sensitive nested PCR and its application for the diagnosis of cutaneous leishmaniasis in Sri Lanka. Microorganisms. 2022;10:990. DOIPubMedGoogle Scholar
Figures
Cite This ArticleOriginal Publication Date: August 13, 2024
1These authors contributed equally to this article.
Table of Contents – Volume 30, Number 9—September 2024
| EID Search Options |
|---|
|
|
|
|
|
|
Please use the form below to submit correspondence to the authors or contact them at the following address:
Nadira D. Karunaweera, University of Colombo, 25, Kynsey Road, Colombo 08, Sri Lanka
Top